R on BioHPC
This guide is a companion to the training sesion 'R on BioHPC' given in July 2015. It introduces:
- The optimized version of R available on BioHPC systems
- The RStudio Server web-based IDE available at rsutdio.biohpc.swmed.edu
- Basic single-node and MPI parallel programming in R
- The Bioconductor bioinformatics package set for R
Why R?
If you are a bioinformatician and regularly look at articles on the web it's almost impossible not to have come across R code, or reference to R. R is an environment for statistics and graphics that has many advantages:
- The dominant statistics environment in academia
- Large number of packages to do a lot of different analyses
- Excellent uptake in Bioinformatics and specialist packages
- (Relatively) easy to accomplish complex stats work
- Very active development right now: R Foundation, R Consortium, Revolution Analytics, RStudio, Microsoft…
Why not R?
Like all languages, R has disadvantages that are important to know before you select it for a project. The most commonly discussed are:
- Quirky language, painful for e.g. Python programmers
- Generally thought to be quite slow, except for optimized linear algebra
- Complex 'old-fashioned' documentation
- Parallelization packages can be complex / outdated
...but it's getting better quickly...
RStudio Server - An IDE for R, on the web
On BioHPC, and elsewhere, the easiest way to get started with R and use it simply and easily is with RStudio, a development and interactive environment for R.
RStudio is available in versions for single computer, as well as a server version that can be installed on a powerful machine and access by many users. At BioHPC we host an RStudio server, accessible via OnDemand RStudio
To use RStudio you need to go to this address and login with your BioHPC username and password. BioHPC RStudio is running on a well-specified virtual server, and can support simultaneous login from many users at one time.
A tutorial on RStudio is outside the scope of this guide. A large amount of information is available on the web, via the RStudio site: Resources on Posit
When to use RStudio
Although the BioHPC RStudio server is a great environment to work with R, it's not suitable for all types of work. As a shared resource on a virtual server it shouldn't be used for very large analyses.
Things you should do on R Studio:
-
Development work with small datasets
-
Creating R Markdown documents
-
Working with Shiny for dataset visualizations
-
Any small, short-running data analysis tasks
Things you should not do on R Studio:
-
Analysis of large datasets requiring many GB of RAM
-
Long running analysis (e.g. multiple hours)
-
Running heavily parallelized code
For these kinds of tasks you must use R on the BioHPC nucleus cluster
Using R on the cluster / clients
If you are logged into the BioHPC cluster or a BioHPC workstation/thin-client it's easy to access our optimized version of R using the module command in a terminal session:
module load R/3.2.1-intelThis gives you access to the latest version of R, optimized, and the same as used by rstudio.biohpc.swmed.edu
Other older versions of R exist as modules, which you can see if you run module avail R. Please only use these versions for consistency in existing projects. The latest version of R should be used for best performance and support.
Installing Packages
We have a set of common packages pre-installed in the R module.
You can install packages into your home directory (~/R) using the standard install.packages command in R. E.g.:
install.packages(c("microbenchmark", "data.table"))Some packages need additional libraries and won’t compile successfully. Ask us to install them for you (biohpc-help@utsouthwestern.edu).
NB This is for packages from CRAN – BioconductoR packages install differently. See later!
BioHPC Optimized R
Our R/3.1.2-intel module was compiled using the Intel 2015 compiler suite, using specific optimizations for the CPUs in our systems and the Intel Math Kernel Library (MKL). The default version of R uses a library for linear algebra that does not make efficient use of modern multi-core CPUs. By using the Intel MKL we can achieve impressive speedups versus a standard R installation:
Task Standard R BioHPC R Speedup
=======================================================================
Matrix Multiplication 139.15 1.80 77x
Cholesky Decomposition 29.53 0.32 61x
SVD 45.66 1.95 23x
PCA 201.30 6.25 32x
LDA 135.37 17.60 7xThis is on a cluster node – speedup is less on clients with fewer CPU cores
For your own Mac or PC see http://www.revolutionanalytics.com/revolution-r-open
Revolution open R contains some of the same optimizations we use, but will run on almost any Windows, Linux or Mac system. BioHPC R is specific to our systems and cannot be used elsewhere.
Benchmarking and Compiling Functions in R
When using any language in an HPC environment it's important to know how to time the execution of portions of your code. In R we generally package important routines into functions, so it's convenient to have an easy way to benchmark and compare the runtime of functions.
The microbenchmark package provides simple function benchmarking, with repeat runs to compute aggregate statistics. We'll use it to look at the effect of compiling a function on its runtime.
As an interpreted language, R translates code into lower level machine code as it runs. This can make it slower that languages such as C, where code is compiled into machine code, and optimized, before a program is run. The compiler package in R allows functions to be compiled to byte-code, so that they run faster when called. This is still slower that compiled C code, but is an easy way to speed up functions which are called many times and/or take a long time to run.
library(compiler)
f <- function(n, x) for (i in 1:n) x = (1 + sin(x))^(cos(x))
g <- cmpfun(f)
library(microbenchmark)
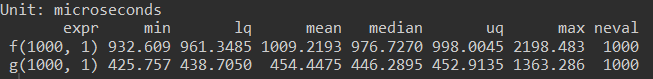
compare <- microbenchmark(f(1000, 1), g(1000, 1), times = 1000)
library(ggplot2)
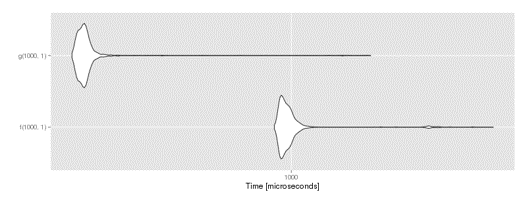
autoplot(compare)In the code above we defined a function f which performs some math in a loop. We translated it into a compiled version g using the cmpfun function from the compiler library. Using microbenchmark we compared f and g, runing each 1000 times. The aggregate statistics can be viewed and plotted:
benchmark results graph
By compiling the function we reduced it's execution time approx 2x. Compilation speedups vary, and are often highest for functions making heavy use of for loops. However, there is a better way to speed up many operations....
For Speed, Always Vectorize!
Like other mathematical environments, the fastest way to perform operations on data in R is generally to use vector/matrix form. Behind the scenes R calls heavily optimized linear algebra libraries. When these libraries are passed vectors and matrices they can compute results far faster than if a for loop is used.
Let's look a function distnorm, which generates X,Y co-ordinates for a plot of the standard normal distribution for -5<x<5. It does this by getting a Y value for each X via the stdnorm function, inside a for loop.
distnorm <- function(){
x <- seq(-5, 5, 0.01)
y <- rep(NA,length(x))
for(i in 1:length(x)) {
y[i] <- stdnorm(x[i])
}
return(list(x=x,y=y))
}Instead of using a for loop we could just pass the vector of X values into stdnorm and receive a vector of Y values back.
vdistnorm <- function(){
x <- seq(-5, 5, 0.01)
y <- stdnorm(x)
return(list(x=x, y=y))
}This simplification results in a 54x speedup
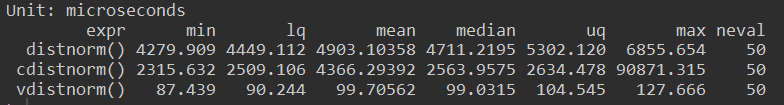
Compiling the function gave a 2x increas in speed, but using vector form is far faster.
Always consider whether for loops in your code can be written to single calls, or multiple steps, on vectors and matrices.
Parallel Programming in R
Most of the time we can rely on the MKL optimizations in BioHPC R and vectorization of our code to keep run time short. The majority of R code will run far more quickly on the BioHPC cluster than your desktop or laptop, and will complete quickly enough that you don't need to worry about parallelizing it. If you do need to parallelize your code, the good news is that is has become easier in R recently, and the same methods apply whether you'll be using a single compute node, or running a large job across our nucleus cluster.
An Example Application for Parallel R
As an example task for our parallel programming section we're going to generate a random walk in 2 dimensions, with a user specified number of steps. Each step will be drawn from a specified normal distbiution, but the bias will decay with each step. Here's the code:
# Define a function that performs a random walk with a
# specified bias that decays
rw2d <- function(n, mu, sigma){
steps=matrix(, nrow=n, ncol=2)
for (i in 1:n){
steps[i,1] <- rnorm(1, mean=mu, sd=sigma )
steps[i,2] <- rnorm(1, mean=mu, sd=sigma )
mu <- mu/2
}
return( apply(steps, 2, cumsum) )
}... and here's an plot of a single walk.
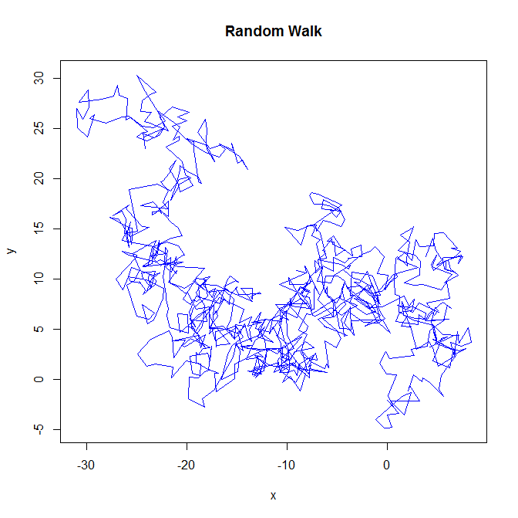
This class of algorithm is sometimes used to explore parameter spaces etc. It's representative of the type of stochastic, iterative procedure that appears a lot in bioinformatics work.
A Bigger Task
Generally with stochastic optimizations or simulations we need to run many replicates and analyze the aggregate output. In this case we will generate 4000 random walks, between 1000 and 5000 steps. We can use a foreach loop or R's apply functions to do this:
# Generate random walks of lengths between 1000 and 5000
# foreach loop
system.time(
results <- foreach(l=1000:5000) %do% rw2d(l, 3, 1)
)
# user system elapsed
# 85.872 0.145 86.242
# Apply
system.time(
results <- lapply( 1000:5000, rw2d, 3, 1)
)
# user system elapsed
# 81.175 0.114 81.511It takes 86s to obtain our 1000 walks using a for loop, or 82s with lapply. Often an apply function is faster than a foreach loop. It's less flexible though, and is less familiar to programmers from other languages.
Single Node Parallelization
Because our 1000 walks are not dependent on eachother we can easily parallelize. This is known as an embarassingly parallel task. Let's try to use all 32 cores available on a single BioHPC compute node, and generate 32 walks at a time.
Recent versions of R include the parallel library, which build on prior work and makes parallelization easy. We load parallel plus doParallel which allows our foreach loop to run in parallel.
#Parallel Single node
library(parallel)
library(doParallel)The parallel package lets us create a cluster of R slave processes. Each of the slave processes can run code at the same time. We usually want to use a slave process (or worker) for each CPU core. We can use the detectCores function so we don't need to hard code the number of cores for each machine.
Once a cluster has been created it must be registered with the doParallel library for parallel foreach loops to work correctly.
# Create a cluster of workers using all cores
cl <- makeCluster( detectCores() )
# Tell foreach with %dopar% to use this cluster
registerDoParallel(cl)
# Parallel loops here
stopCluster(cl)We call stopCluster to shut down our worker processes when we are finished running our parallel code. The workers will stop automatically when we quit R, but until then they will use RAM unless we shut them down explicitly.
R Parallel vs MKL Conflict
Before we run parallel code it's important to be aware of a conflict between our optimize MKL version of R, and explicit parallelization with the parallel package.
Intel MKL tries to use all cores for every linear algebra operation. When we use the parallel package, R is running multiple iterations of a loop in parallel using all cores.
If used together too many threads/processes are launched – far more than cores!
We can disable the MKL multi-threading from the command line before starting R:
export OMP_NUM_THREADS=1 # on terminal before running R... or from inside R, in our scripts etc.
sys.setenv(OMP_NUM_THREADS="1") # within RThere's generally a ~5% improvement by disabling MKL multi-threading, if our code is using explicit parallelization efficiently. In some cases, when your parallelization is incomplete or inefficient, it may be better to leave MKL multi-threading on. Test and time your code to find the best choice.
Our 2D Walks in Parallel
Back to our 2D walks, let's now generate them in parallel. We just change our foreach loop from %do% to %dopar, and our apply function from lapply to parLapply.
cl <- makeCluster( detectCores() )
RegisterDoParallel(cl)
Sys.setenv(OMP_NUM_THREADS="1")
# Generate 1000 random walks of increasing length
# Parallel foreach loop
system.time(
results <- foreach(l=1000:5000) %dopar% rw2d(l, 3, 1)
)
# user system elapsed
# 2.928 0.441 17.374
# Parallel apply
system.time(
results <- parLapply( cl, 1000:5000, rw2d, 3, 1)
)
# user system elapsed
# 0.339 0.171 8.460
stopCluster(cl)So, That's a 5x speedup for the foreach loop, and a 9x speedup for the lapply. Not bad!
We have 32 cores, but we don't see a 32x speedup as we were already benefitting from some MKL multi-threading in the sequential code, and there is overhead communicating tasks and results between our parallel worker processes.
MPI Parallelization - For Really Big Jobs
When a task is so complex that a single node can't finish an analysis in reasonable time you need to run code across multile nodes in the nucleus compute cluster. Our cluster has a fast interconnect, which makes passing messages between nodes efficient. We use the MPI framework to run code in parallel across the cluster, passing messages for co-ordinations.
MPI is available on R/3.1.2-intel only
We will continue to use the simple parallel and doParallel packages. If you search the web you'll find Lots online about ‘snow’ – this package is now behind the scenes in new versions of R, hidden by the simple parallel package.
Please let us know if you use MPI in R. We're keen to take a look at the types of problem you are tackling, help you optimize code, and optimize our configuration for best performance.
Enabling MPI in R - Easy!
Writing MPI programs from scratch in other languages is difficult. Luckily in R all we need to do is tell the parallel package to make and MPI cluster.
Just one change in our R code!
cl <- makeCluster( 128, type="MPI" )Here, 128 is the number of MPI tasks, and should be:
cores per node * nodes (or less if RAM limited)On our systems we have:
- 48 cores per node for 256GB partition
- 32 cores per node for other partitions
MPI Job Script
MPI R code can't be launched from within a standard R session on a single machine. Because we will be running our code across multiple nodes in the compute cluster we must create a job script and submit it to the SLURM scheduler.
Here's the script to launch our MPI R code, assuming it's in the fie named mpi_parallel.R
#!/bin/bash
#SBATCH --job-name R_MPI_TEST
# Number of nodes required to run this job
#SBATCH -N 4
# Distribute n tasks per node
#SBATCH --ntasks-per-node=32
#SBATCH -t 0-2:0:0
#SBATCH -o job_%j.out
#SBATCH -e job_%j.err
#SBATCH --mail-type ALL
#SBATCH --mail-user david.trudgian@UTSouthwestern.edu
module load R/3.2.1-intel
ulimit -l unlimited
R --vanilla < mpi_parallel.R
# END OF SCRIPTWe can submit the job using the command
sbatch mpi_parallel.sh...and use the squeue command to see the job status.
MPI Performance
Our simple MPI script is computing 9000 2D random walks, more than the single-node code in order that we can see the MPI speedup. It takes time to build up the MPI R workers, and there is overhead transmitting data between them,
# Sequential (with MKL multi-threading)
system.time(
results <- lapply( 1000:10000, rw2d, 3, 1)
)
# user system elapsed
# 329.173 0.610 330.607Running the 9000 walks sequentially on a single node (allowing MKL multi-threading) requires 330s.
# Parallel apply, 4 nodes, 128 MPI tasks
system.time(
results <- parLapply( cl, 1000:10000, rw2d, 3, 1)
)
# user system elapsed
# 18.815 0.951 19.848Running in parallel across 4 nodes decreases the elapsed time to 20s, for a 16x speedup. This is quite high for such a simple task. The largest speedup will be seen on tasks that perform a lot of computation and make small, infrequent data transfers.
Bioconductor
Our final topic is Bioconductor, a comprehensive suite of packages for bioinformatics in R. Bioconductor contains both software and datasets, and is a great resource for anyone working with any biological data - sequences, images, clinical outcomes etc.
Our R module has Bioconductor base packages installed, plus some commonly used extras
You can install additional Bioconductor packages to your home directory:
source("http://bioconductor.org/biocLite.R")
biocLite('limma')Ask biohpc-help@utsouthwestern.edu for packages that fail to compile
Follow our separate Bioconductor guide to learn more.