Matlab 2014b and above can experience the following issues when run on BioHPC systems. BioHPC has reported these issues to the MATLAB developer, but no fix is currently available.
- The built-in `mkdir` function to create directories gives a 'Permission Denied' error on the /project directory.
Please use the 'system('mkdir -p mydir')' command instead of mkdir.
- If editor sessions were open when MATLAB was closed, it will fail to load with a library error when next run on a webGUI or thin-client session.
Please start MATLAB with the -softwareopengl option 'matlab -softwareopengl' as a workaround.
Matlab 2018b job submission to the cluster can fail if ran directly from a workstation or a thinclient. As a workaround, consider
- Running Matlab 2018b directly from a WebGUI session
- Using Matlab 2019a/b
MATLAB Parallel Computing on BioHPC
- Build-in Multithreading
Automatically handled by MATLAB, use of vector operations in place of
forloops. - Parallel Computing Toolbox
Acceleration MATLAB code with very little code changes, job will running on your workstation, thin-client or a reserved compute node
- Matlab Distributed Computing Server (MDCS)
- Directly submit matlab job to BioHPC cluster
- MATLAB job scheduler integrated with
SLURM
Parallel Computing Toolbox
Parallel for-Loops (parfor)
- Allow several MATLAB workers to execute individual loop iterations simultaneously
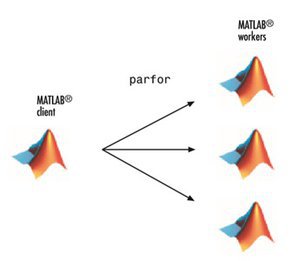
- The only difference in parfor loop is the keyword
parforinstead offor. When the loop begins, it opens a parallel pool of MATLAB sessions called workers for executing the iterations in parallel.
Example: Estimating an Integration
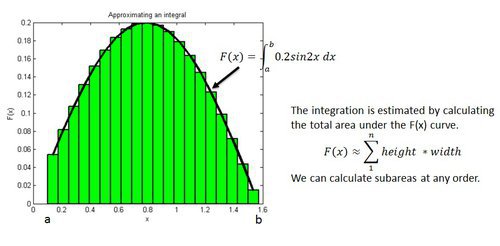
Implementation
function q = quad_fun (n, a, b)
q = 0.0;
w=(b-a)/n;
for i = 1 : n
x = (n-i) * a + (i-1) *b) /(n-1);
fx = 0.2*sin(2*x);
q = q + w*fx;
end
return
end
run from command line:
>> tic
q=quad_fun(120000000, 0.13, 1.53);
t1=toc
t1 =
5.7881
function q = quad_fun_parfor (n, a, b)
q = 0.0;
w=(b-a)/n;
parfor i = 1 : n
x = (n-i) * a + (i-1) *b) /(n-1);
fx = 0.2*sin(2*x);
q = q + w*fx;
end
return
end
run from command line :
>> parpool('local');
Starting parallel pool(parpool) using the 'local' profile ... connected to 12 workers ...
>> tic
q=quad_fun_parfor(120000000, 0.13, 1.53);
t2=toc
t2 =
0.9429
12 workers, Speedup = t1 / t3 = 8.26x
Limitations of parfor
- No Nested parfor loops
- Loop variable must be increasing integers
- Loop iterations must be independent
spmd
General
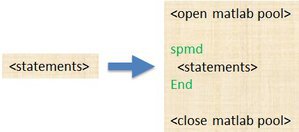
- single program -- the identical code runs on multiple workers.
- multiple data -- each worker can have different, unique data for that code.
- numlabs – number of workers.
- labindex -- each worker has a unique identifier between 1 and numlabs.
- point-to-point communications between workers : labsend, labreceive, labsendrecieve
- Ideal for: 1) programs that takes a long time to execute. 2) programs operating on large data set.
spmd
labdata = load(['datafile_' num2str(labindex) '.ascii']) % multiple data
result = MyFunction(labdata); % single program
end
Example: Estimating an Integration
quad_fun_spmd.m (spmd version)
a = 0.13;
b = 1.53;
n = 120000000;
spmd
aa = (labindex - 1) / numlabs * (b-a) + a;
bb = labindex / numlabs * (b-a) + a;
nn = round(n / numlabs)
q_part = quad_fun(nn, aa, bb);
end
q = sum([q_part{:}]);
run from command line :
8.26x
>> tic
quad_fun_spmd
t3=toc
t3 =
0.7006
Q: why better performance compares to parfor version ?
A: 12 communications between client and workers to update q, whereas in parfor, it needs 120,000,000 communications between client and workers.
Distributed Array
- Distributed Array - Data distributed from client and access readily on client. Data always distributed along the last dimension, and as evenly as possible along that dimension among the workers.
- Codistributed Array – Data distributed within spmd. Array created directly (locally) on worker.
Examples
Matrix Multiply (A*B) by Distributed Array
parpool('local');
A = rand(3000);
B = rand(3000);
a = distributed(A);
b = distributed(B);
c = a*b; % run on workers automatically as long as c is of type distributed
delete(gcp);
Matrix Multiply (A*B) by Distributed Array
parpool('local');
A = rand(3000);
B = rand(3000);
spmd
u = codistributed(A, codistributor1d(1)); % by row
v = codistributed(B, codistributor1d(2)); % by column
w = u * v;
end
delete(gcp);
Linear solver (Ax = b)
Implementation
n = 10000;
M = rand(n);
X = ones(n, 1);
A = M + M';
b = A * X;
u = A \ b;
run from command line :
>>tic;linearSolver;t1=toc
t1 =
8.4054
n = 10000;
M = rand(n);
X = ones(n, 1);
spmd
m = codistributed(M, codistributor('1d', 2)); % distribute one portion of m to each MATLAB worker
x = codistributed(X, codistributor('1d', 1)); % distribute one portion of x to each MATLAB worker
A = m + m';
b = A * x;
utmp = A \ b;
end
u1 = gather(utmp); % gathering u1 from all MATLAB workers to MATLAB client
run from command line :
>>parpool('local');
Starting parallel pool(parpool) using the 'local' profile ... connected to 12 workers
>>tic;linearSolverSpmd;t2=toc
t2 =
16.5482
12 workers, Speedup = t1 / t3 = 8.26x
Q: why spmd version needs more time for job completion ?
A: It needs extra time for distribute/consolidate large data and tranfer them to/from different workers
Factors reduce the speedup of
parfor and spmd:
- Computation inside the loop is simple.
- Memory limitations. (create more data compare to serial code)
- Transfer data is time consuming
- Unbalanced computational load
- Synchronization
Contrast Enhancement

contract_enhance.m
function y = contrast_enhance (x)
x = double (x);
n = size (x, 1);
x_average = sum ( sum (x(:, :))) /n /n ;
s = 3.0; % the contrast s should be greater than 1
y = (1.0 – s ) * x_average + s * x((n+1)/2, (n+1)/2);
return
end
Implementation
% contrat_serial.m
x = imread('surfsup.tif');
yl = nlfilter (x, [3,3], @contrast_enhance);
y = uint8(y);
% contrat_parallel.m
x = imread('surfsup.tif');
xd = distributed(x);
spmd
xl = getlocalPart(xd);
xl = nlfilter(xl, [3,3], @contrast_enhance);
end
y = [xl{:}];
12 workers, running time is 2.01 s, Speedup = 7.19x

Problem : When the image is divided by columns among the workers, artifcial internal boundaries are created !
Reason : Zero padding at edges when applying sliding-neighborhood operation.
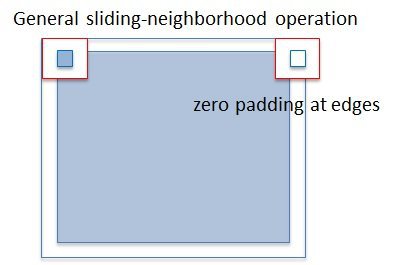
Solution: Build up communication between workers.
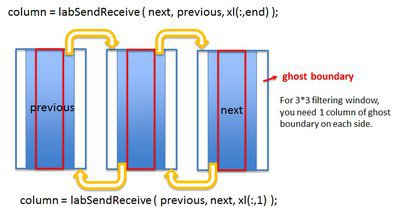
contrast_MPI.m
x = imread('surfsup.tif');
xd = distributed(x);
spmd
xl = getLocalPart ( xd );
% find out previous & next by labindex
if ( labindex ~= 1 )
previous = labindex - 1;
else
previous = numlabs;
end
if ( labindex ~= numlabs )
next = labindex + 1;
else
next = 1;
end
% attach ghost boundaries
column = labSendReceive ( previous, next, xl(:,1) );
if ( labindex < numlabs )
xl = [ xl, column ];
end
column = labSendReceive ( next, previous, xl(:,end) );
if ( 1 < labindex )
xl = [ column, xl ];
end
xl = nlfilter ( xl, [3,3], @contrast_enhance );
% remove ghost boundaries
if ( 1 < labindex )
xl = xl(:,2:end);
end
if ( labindex < numlabs )
xl = xl(:,1:end-1);
end
xl = uint8 ( xl );
end
y = [ xl{:} ];
12 workers, running time is 2.01 s, Speedup = 7.23x
pmode
pmode allows the interactive parallel execution of MATLAB® commands. pmode achieves this by defining and submitting a communicating job, and opening a Parallel Command Window connected to the workers running the job. The workers then receive commands entered in the Parallel Command Window, process them, and send the command output back to the Parallel Command Window. Variables can be transferred between the MATLAB client and the workers.
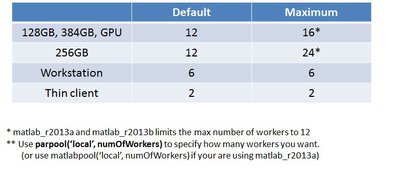
Q: How many workers can I use for MATLAB parallel pool ?
GPU
Parallel Computing Toolbox enables you to program MATLAB to use your computer's graphics processing unit (GPU) for matrix operations. For some problems, execution in the GPU is faster than in CPU.
To enable GPU computing of MATLAB on BioHPC cluster:
- reserve a GPU node by remoteGPU or webGPU
- inside terminal, type in
export CUDA_VISIBLE_DEVICES=“0”
You can lauch Matlab directly if you have GPU card on your workstation.
linear Solver (Ax = b) using GPU
n = 10000; M = rand(n); X = ones(n, 1); % Copy data from RAM to GPU ( ~ 0.3 s) Mgpu = gpuArray(M); Xgpu = gpuArray(X); Agpu = Mgpu + Mgpu'; Bgpu = Agpu * Xgpu; ugpu = Agpu \ bgpu % Copy data from GPU to RAM ( ~0.0002 s) u = gather(ugpu);
run from command line :
>>tic;linearSolverGPU;t3=toc t3 = 3.3101
Speedup = t1 / t3 = 2.54x


Parallel Computing Toolbox – Benchmark
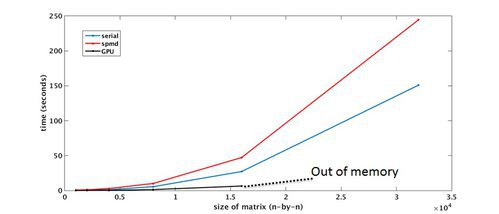
MATLAB Distributed Computing Server (MDCS)
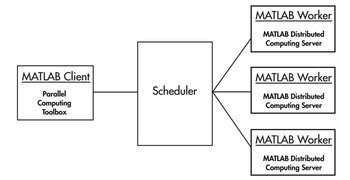
See below for configuring your MATLAB profile to communicate with the BioHPC cluster
- Open 'MATLAB Cluster Profile' manager
Home -> ENVIRONMENT -> Parallel -> Manage Cluster Profiles
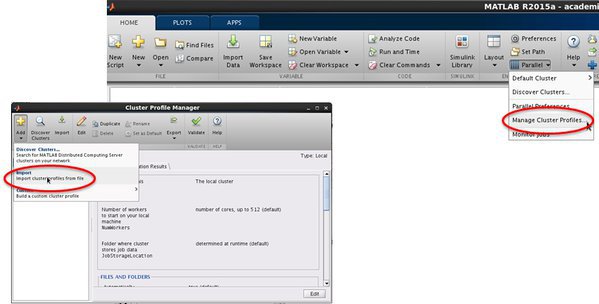
- Import a cluster profile setting
And select a profile on the cluster:
Add -> Import Cluster Profiles from file -> Import cluster profile from file
/project/apps/MATLAB/profile/nucleus_r2016a.settings/project/apps/MATLAB/profile/nucleus_r2015b.settings/project/apps/MATLAB/profile/nucleus_r2015a.settings/project/apps/MATLAB/profile/nucleus_r2014a.settings/project/apps/MATLAB/profile/nucleus_r2014b.settings/project/apps/MATLAB/profile/nucleus_r2013b.settings/project/apps/MATLAB/profile/nucleus_r2013a.settings
- Test MATLAB environment (optional)
After initial setup, you should have two Cluster Profiles ready for Matlab parallel computing:
“local" – for running job on your workstation, thin client or on any single compute node of BioHPC cluster (use Parallel Computing toolbox)
"nucleus_r<version No.>" – for running job on BioHPC cluster with multiple nodes (use MDCS)
Setup Slurm Environment from Matlab Command line
ClusterInfo.setQueueName('128GB'); % use 128 GB partition
ClusterInfo.setNNode(2); % request 2 nodes
ClusterInfo.setWallTime('1:00:00'); % setup time limit to 1 hour
ClusterInfo.setEmailAddress('yi.du@utsouthwestern.edu'); % email notification
Check Slurm Environment from Matlab Command line
ClusterInfo.getQueueName();
ClusterInfo.getNNode();
ClusterInfo.getWallTime();
ClusterInfo.getEmailAddress();
- Open 'MATLAB Cluster Profile' manager
Home -> ENVIRONMENT -> Parallel -> Manage Cluster Profiles
- Add a Slurm cluster
Add -> Slurm
- Edit the profile, and set partition name under 'Additional command line arguments for job submission :
--partition=super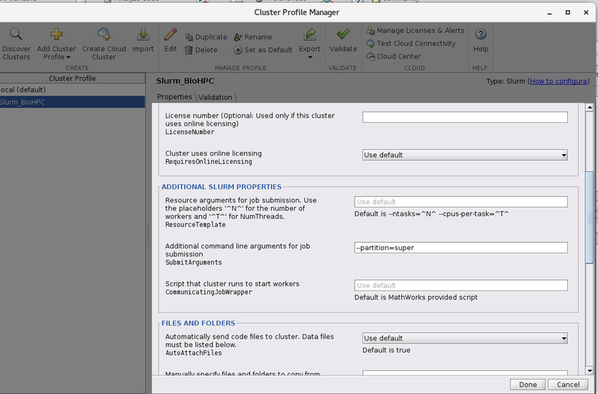
- Test MATLAB environment (optional)

wait(job, 'finished'); % Block session until job has finished get(job, 'State'); % Occasionally checking on state of job
results = fetchOutputs(job); % load job results results = load(job); % load job results, only valid when submit job as a script delete(job); % delete job related data
You can also open job monitor from Home->Parallel->Monitor Jobs
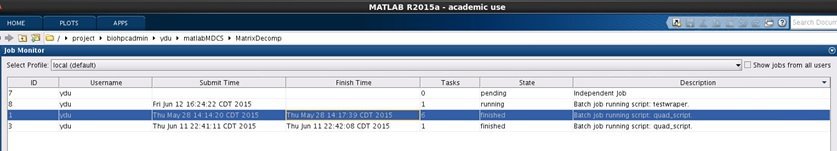
Examples
Estimating an Integration
% setup Slurm environment ClusterInfo.setQueueName('super'); ClusterInfo.setNNode(2); ClusterInfo.setWallTime('00:30:00'); % submit job to BioHPC cluster job = batch(@quad_fun, 1, {120000000, 0.13, 1.53}, 'Pool', 63, 'profile', 'nucleus_r2015a'); % wait wait(job, 'finished'); % load results after job completion Results = fetchOutputs(job);
biohpc_cluster = parcluster('NAME_OF_CREATED_SLURM_PROFILE'); % Alternatively, create on for current session: % biohpc_cluster = parallel.cluster.Slurm; % Optional -- Specify/overwrite additional Slurm arguments % biohpc_cluster.SubmitArguments = "--nodes=4" ... % + " --time=24:00:00" + ... % + " --hint=nomultithread"; % submit job to BioHPC cluster job = batch(biohpc_cluster, @quad_fun, 1, {120000000, 0.13, 1.53}, 'Pool', 63); % wait wait(job, 'finished'); % load results after job completion Results = fetchOutputs(job);
check Slurm settings from Matlab command window

check Job status from terminal ( squeue -u <username>)

load results
16 workers cross 2 nodes, running time is 24s
Recall that the serial job only needs 5.7881 seconds, MDCS needs more time for Example 1 as it is not designed for small jobs.
Scaling and Application: Vessel Extraction
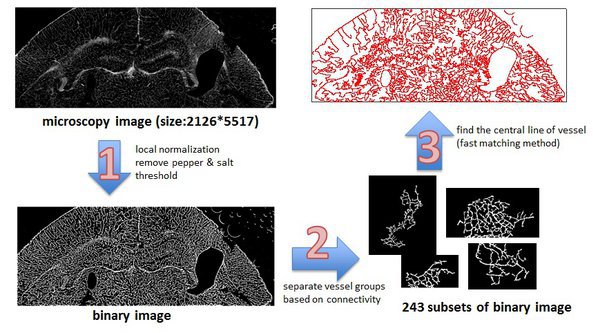
parfor i = 1 : length(numSubImages) I = im2double(Bwall(:,:i)); % load binary image allS{i} = skeleton(I); % find vessel by using fast matching method end
performance evaluation ( running time v.s. number of workers/nodes)
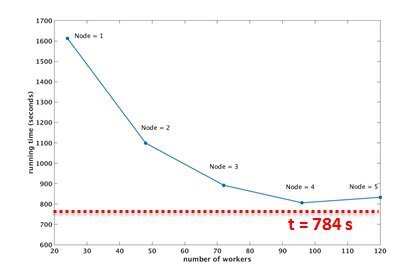
Job running on 4 nodes (96 workers) requires minimum computational time, Speedup = 28.3 x @ 4 nodes.
time needed for each single image various
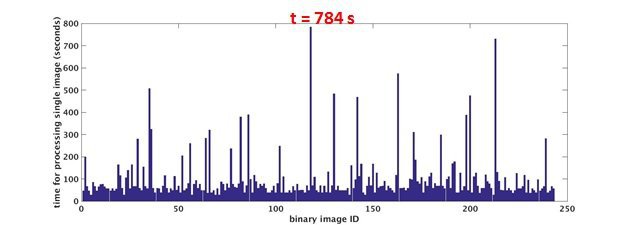
Why linear speedup is impossible to obtain ?
- overhead caused by load balancing, synchronization, communication and etc..
- limited by the longest (single/serial) job