Galaxy on BioHPC
Introduction
The BioHPC Galaxy service is BioHPC's installation of Galaxy - an open-source multi-institution project to provide a platform for reproducible analysis of large datasets. Galaxy is a web-based system that provide tools (which analyze data), in an environment which maintains histories of analyses run, and the ability to define workflows. The Galaxy project maintains a website at: http://www.galaxyproject.org which holds various introductory and technical documentation. The project hosts a public Galaxy service at http://usegalaxy.org but this provides limited working space, is inappropriate for private data, and job wait times can be high.
The BioHPC Galaxy service allows any BioHPC user to use Galaxy, internally on BioHPC systems. The service is configured to store data on BioHPC's large storage systems and use the Nucleus cluster for long-running analysis jobs.
The BioHPC galaxy service is located at: http://galaxy.biohpc.swmed.edu
Important: Another group on campus, QBRC, runs a galaxy server at galaxy.swmed.edu. Ensure you are using the BioHPC galaxy.biohpc.swmed.edu server when following these notes.
How to Learn Galaxy
Galaxy is a large and complex system, one that is extremely powerful and flexible for NGS analyses. The training session can only give an extremely quick introduction to the system. If Galaxy proves popular we are likely to offer an additional session. Until then, there are a lot of resources on the web that introduce Galaxy, and you can attend the BioHPC coffee sessions or contact us to request help with analyses.
We strongly advise working through the material on the UseGalaxy wiki at:
https://wiki.galaxyproject.org/Learn
A number of screencasts and demos are available, and are highly recommended:
https://wiki.galaxyproject.org/Learn/Screencasts
Other institutions offer specific training material that may be valuable, but will usually include specifics for their Galaxy Service. We hope to provide extensive tutorials customized for BioHPC in future. However, you may wish to look at the following material from other Universities:
http://training.bioinformatics.ucdavis.edu/docs/2014/12/december-2014-workshop/
http://www.slideshare.net/afgane/introduction-to-galaxy-and-rnaseq
This document gives a very brief introduction to some key concepts, and highlights things that are specific to the BioHPC Galaxy installation. Please read these sections carefully,
Login, Groups, Sharing
The BioHPC galaxy service is available to registered users only. You must login at galaxy.biohpc.swmed.edu using your BioHPC username and password.
Galaxy maintains its own structure of user groups and roles. Any work you do in Galaxy is not automatically accessible by others and Galaxy does not know about lab and departmental groups.
If you wish to share data with other users in Galaxy you should contact biohpc-help at utsouthwestern dot edu for assistance. We hope to modify Galaxy in future so that it understands our department / lab group structure for easy collaboration within these groups.
Basic Concepts - Histories, Tools, Workflows
Galaxy is designed to support reproducible research. It is structured so that it keeps track of all the actions that are run in a dataset so that these can be reviewed or repeated in future. The following concepts support this way of working:
Histories - In Galaxy all work is done within a history. A history contains associated datasets, analyses run on the data, and outputs from the analyses. Any time you are logged into Galaxy you are working in your current history. The current history is displayed in the right side-bar on the Galaxy web interface. As you upload data, run tools etc. items will be added to the history. Each user can create unlimited histories. You can name a history, and choose to keep it for reference. If you are just trying things out you can delete the history when you are done.

Histories are convenient for other reasons - for publication you may export a history, so that you have an exact record of the analysis run in an experiment. You can also export a list of citations for a history, so you can easily cite tools that were used in your analysis.
Read the Galaxy documentation on histories here: https://wiki.galaxyproject.org/Histories
Tools - In Galaxy, a tool is something that can run on some input data and produce output. Galaxy is a modular system - many different developers create tools that can be installed onto the Galaxy server. Most tools are wrappers around a piece of bioinformatics software. E.g. there are tools which run the well-known bowtie aligner on short-read sequencing datasets.
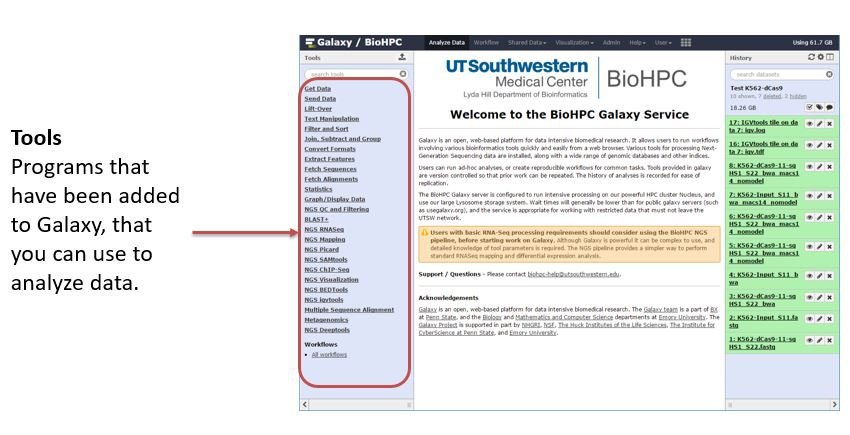
Tools are listed in categories in the tool panel at the left side of the window. If you select a tool you will see a screen of options, where you choose input data, tool parameters, and can submit the analysis job. Not all tools available to Galaxy are installed on our system, as each tool takes time to configure and must be kept up-to-date. There is a public toolshed at https://toolshed.g2.bx.psu.edu/ with a list of tools available. You can request installation of tools by emailing biohpc-help@utsouthwestern.edu
Workflows - Although you can use Galaxy in a step-by-step manner, running each tool in a pipeline manually, this become tedious for large analyses. Workflows allow you to create your own pipelines, linking the output of one tool to the input of another so that a complete analysis can be run with one action. BioHPC will try to generate public workflows as templates for common tasks. Please read the Galaxy workflow documentation online which explains how to use this powerful feature:
https://wiki.galaxyproject.org/Learn/AdvancedWorkflow
Uploading Data - From the Browser
The easiest way to upload small datasets to Galaxy is via your web browser. Click the upload data button at the top of the tool bar.
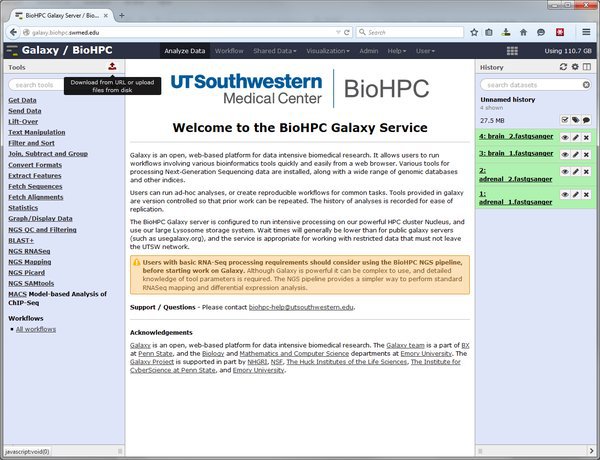
In the upload dialog click the 'Choose local file' button and select the file(s) to upload from your local computer. When you have selected your file set the type and genome for each file, and click the Start button to begin the upload.
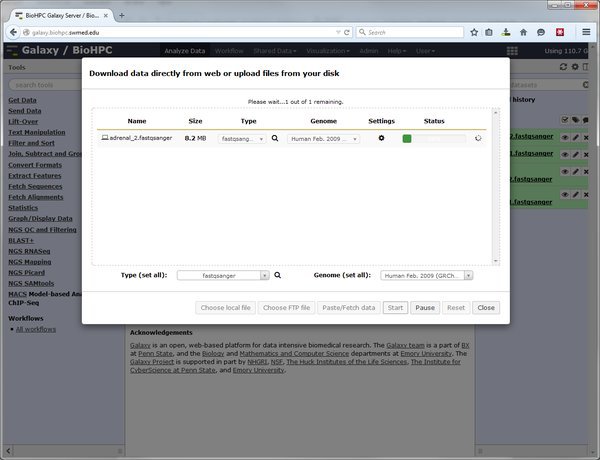
LIMITATIONS - Files >4GB cannot be uploaded from most web browsers. Due to the configuration of Galaxy the practical limit for file uploads is ~200MB. Beyond this please use the alternative method of importing data from the cluster.
Uploading Data - Importing from galaxy_incoming
Many Galaxy tutorials will talk about 'FTP upload' of datasets. This is a function of Galaxy which allows it to receive arbitrarily large files that cannot be uploaded via the web browser. In the BioHPC setup, Galaxy is not used to manage FTP connections and accounts. FTP upload does not work in the same way as public Galaxy installations on the web.
To import large files in Galaxy you must copy them to a special location on cluster storage. This location is:
/project/apps/galaxy/galaxy_incoming/<username>
... where <username> should be replaced with your BioHPC username.
Copy any file you want to use in Galaxy into this directory on the cluster storage. DO NOT link or move files to here. Galaxy deletes files in the incoming folder as it imports them.
You can copy files to this location in various ways:
-
Directly copying from another location on /home /project /work using a BioHPC workstation, a nucleus terminal session etc.
-
Using an FTP client, connecting to lamella.biohpc.swmed.edu and uploading files to the appropriate path.
-
Using the lamella cloud storage interface. Mount your galaxy_incoming directory into the lamella interface and copy or upload files via lamella.
Once you have copied files into your galaxy_incoming directory you import them via the Galaxy web interface. Click the upload data button, and choose the 'Choose FTP file' button. This will open a list of all files in your galaxy_incoming directory which are available to import:
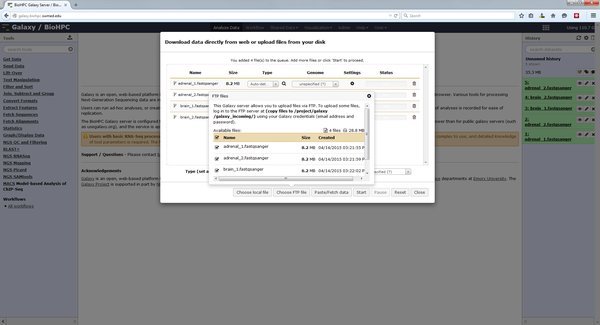
Select the files to import into your current Galaxy history, close the FTP files window, and Start the import process. The files will be imported into Galaxy.
FILES IN YOUR GALAXY_INCOMING DIRECTORY ARE DELETED AS GALAXY IMPORTS THEM! MAKE SURE THAT THEY ARE ONLY A COPY. DO NOT MOVE FILES TO THIS LOCATION.
Uploading Data - Direct download from the web
If you are using public datasets that are available from the internet on a public site you can ask Galaxy to download them directory into your history. This is possible via the Paste/Fetch data option in the upload dialog. Paste a list of HTTP or FTP URLs to the data you want to analyze with Galaxy:
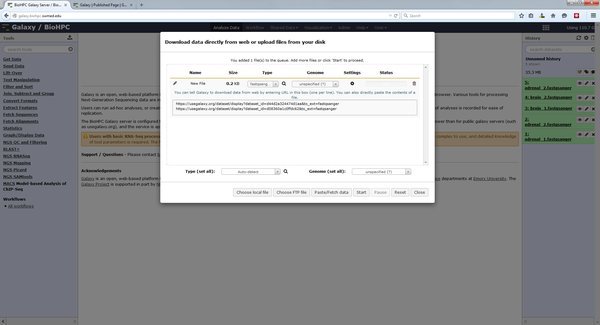
When you press start Galaxy will create jobs in your history that will fetch the files specified from the internet. This can be useful if you want to download a large set of public data. The galaxy server has a faster connection to the internet than many computers on campus.
Running a Galaxy Tool on Your Data
Once you have added data to your Galaxy history you are ready to perform analysis by running tools against the data. A list of tools is found on the left side of the galaxy main screen. When you click on a tool name options for that tool will appear in the center pane:
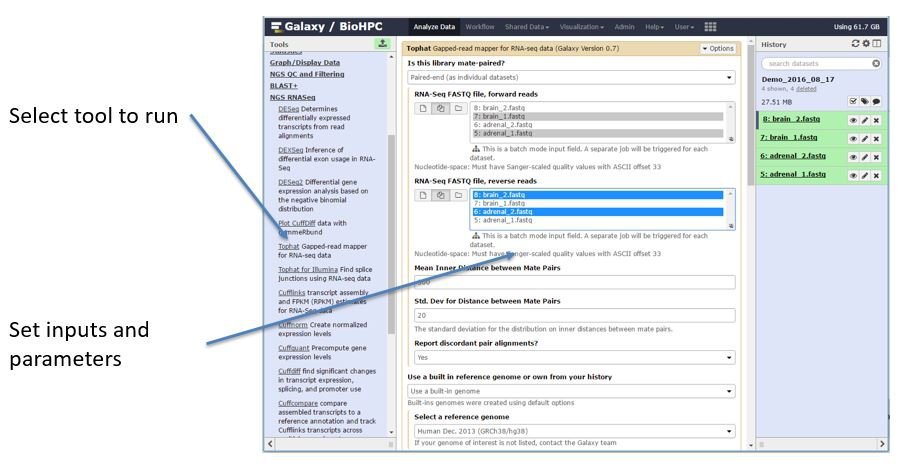
Most tools support selecting one input, or colllections of multiple inputs. If you use a collection of multiple inputs the tool will run on each input. Parameters for the tool are presented in sections, with the advanced parameters hidden at the bottom of the form. When you have chosen the parameters you need and click to run the tool you will be shown a screen which lists the outputs that the tool will generate. These outputs will also be listed in the history:
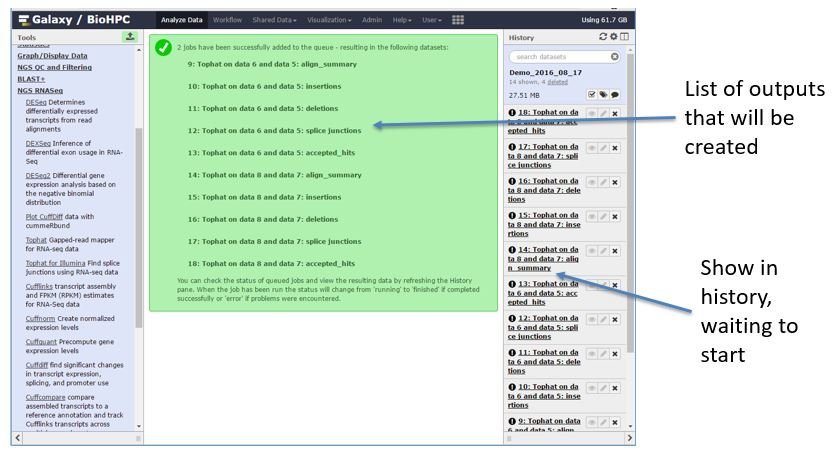
If a tool generates different types of output files then there will be multiple history entries. The entries begin grey as the run is being prepared, turn yellow as it starts, then to green when complete.
![]()
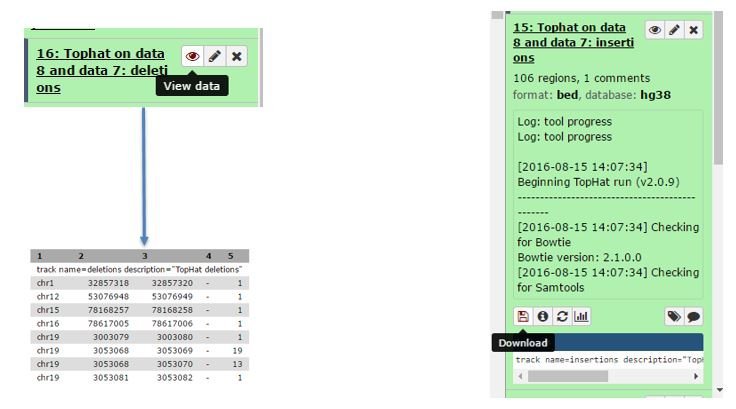
Viewing and Downloading Results
Some types of outputs can be viewed in Galaxy by clicking the eye icon associated with an output in the history bar. Other outputs can only be downloaded to view or work with on your computer:
Workflows
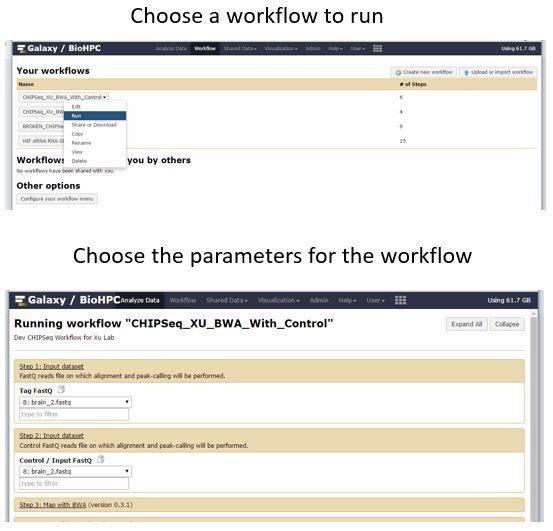
When you are ready to move on from running individual tools to more automated analysis you can start to use and create Galaxy workflows. The 'Workflows' link in the top menu bar gives access to the workflow functionality of Galaxy. A workflow links together multiple tools so that the entire process can be launched from a single form. Workflows can be shared. BioHPC has made available some example ChipSeq workflows which you can run against your data, or test-data as an exercise. To run a workflow select it from the 'Your workflows' section - click the drop down menu and choose Run. The center pane will be replaced with a form to set the parameters of the workflow. The workflow designer decides how many parameters to allow you to tune for each step of the workflow. Others are set to defaults - but can be changed here if needed. When you run a workflow against input all the workflow steps will be added to a history, so you can monitor progress and view or download results.
Designing Workflows
You can create workflows in two ways - by either extracting a workflow from a history you have created manually, or using the visual workflow designer. The workflow designer allows you to add and link tools together using a drag-and-drop interface.
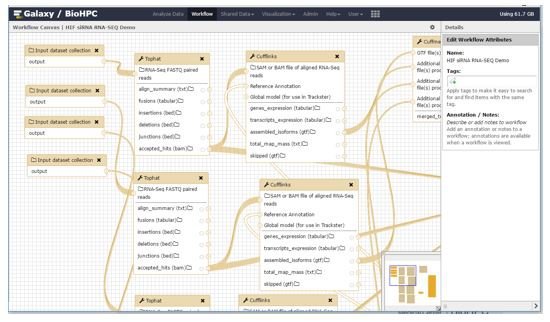
Documentation for workflow design is outside of the scope of this guide. The Galaxy developers provide many guides, screencasts and examples at: https://wiki.galaxyproject.org/Learn/AdvancedWorkflow
Job Submission & Execution
When you submit a job in galaxy, by running a tool on some input data, a handler decides where that job will run. The BioHPC Galaxy service is configured to run small jobs locally, on the Galaxy server itself, so you don't have to wait for free slots on the Nucleus cluster. Tools which generally require a lot of processing power, memory, or run for a long time are configured to run on the cluster. These jobs are run under your own user account, so compute usage is tracked as if you submitted the job manually.
We will continually adjust the balance of local vs cluster jobs depending on the workload of the system. If you find that excessive wait times are interrupting your workflow please email us via biohpc-help@utsouthwestern.edu as it's possible to customize the scheduling of jobs more finely once we have an idea of usage.
Storage & Quotas
At present, disk usage within Galaxy does not count toward your group quota. However, we intend to change this in future. In the short term we will monitor usage, and may need to impose separate quotas within Galaxy if it is excessive. You can always see how much space you are using with Galaxy at the top-right of the web interface. Note that delete histories are not purged
Note - There is no access to any data in Galaxy except via the web interface. Galaxy does not store datasets in a traditional file structure - you must download files from your Galaxy history on the web if you need to use them elsewhere.